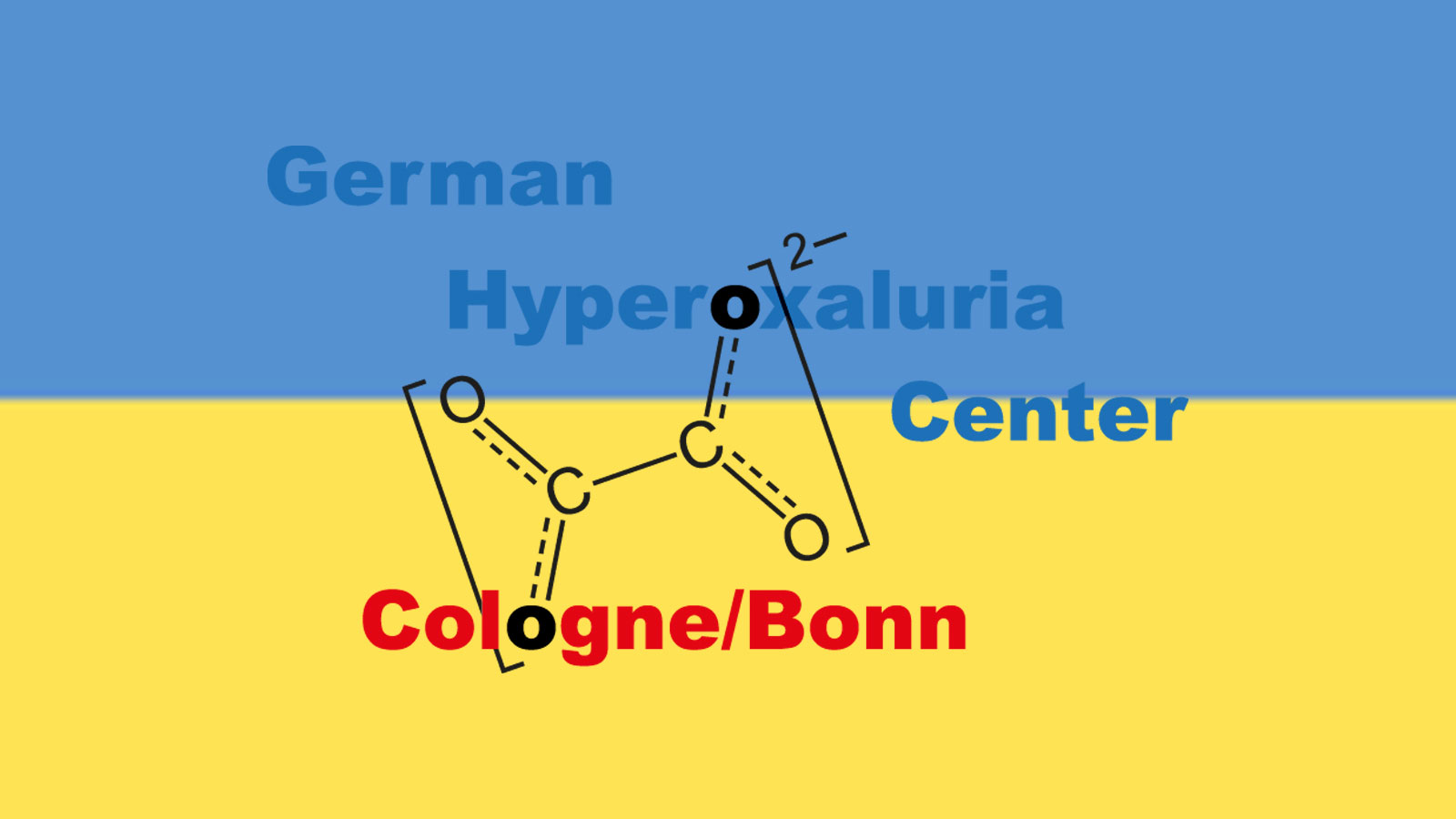
An increased urinary excretion of oxalate is defined as hyperoxaluria, which always induces supersaturation of the urine with regard to calcium-oxalate (CaOx). Such supersaturation leads to CaOx crystal aggregation and later to either recurrent kidney stones or calcium-oxalate deposition within the kidney parenchyma = oxalosis.
Three primary are differentiated from the secondary forms of hyperoxaluria. A primary form is defined as endogenous overproduction of oxalate based on a defined enzyme defect within the glyoxylate metabolism. Currently three forms are known, of which primary hyperoxaluria type I is the most often and the most dramatic form. Here, early end stage renal failure is often the case and even infantile forms with renal failure within the first months of life are known. However, PH types II and III can also lead to severe recurrent kidney stone disease or progressive calcium-oxalate deposition within the kidney.
Secondary hyperoxaluria is frequently found in patients with chronic intestinal bowel diseases (Crohn's), or malabsorption syndromes (celiac disease, cystic fibrosis), or short bowel syndrome. In all this diseases intestinal oxalate absorption is exaggerated, which later has to be excreted via the kidneys. Also, increased dietary intake of oxalate or absence of intestinal oxalate degrading bacteria can lead to oxalate overabsorption and later to hyperoxaluria.
Therefore, every patient with calcium-oxalate stones or nephrocalcinosis/oxalosis has to be screened for hyperoxaluria. We can offer this diagnostic evaluation starting with the analysis of urinary constitutents, plasma oxalate determination and later, if a primary hyperoxaluria is suspected with mutation analysis of the specific gene.
Besim is one of these patients. He has primary hyperoxaluria type I, an ultra-rare disease of the liver, but one that first leads to kidney problems as it progresses. But, let's let Besim and his mother explain his medical history..
Contact us
Info@hyperoxalurie-zentrum.de
Deutsches Hyperoxaluriezentrum, c/o Kindernierenzentrum Bonn, Dr. Gesa Schalk & Prof. Dr. Bernd Hoppe, Im Mühlenbach 2b, 53127 Bonn